

Neue DNA-Sequenzierungstechnologien und Algorithmen zur Sequenzanalyse in der Bioinformatik ermöglichen die Erstellung hochwertiger Chromosomen-Assemblierungen großer Genome. Viele Forschungsgemeinschaften stützen sich jedoch nach wie vor auf fragmentierte, unvollständige und ohne Chromosomenortungsdaten versehene Assemblierungsentwürfe der „Version 1.0“. In einer in der Fachzeitschrift BMC Biology veröffentlichten Studie zeigt die SIB-Gruppe um Robert Waterhouse (Universität Lausanne), wie genomvergleichende evolutionäre Ansätze genutzt werden können, um solche Entwürfe auf ihrem Weg zu „fertigen“ Referenzgenomen zu unterstützen.
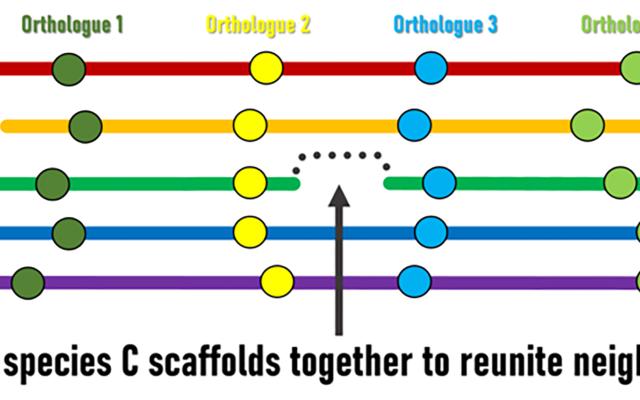
Ausnutzung konservierter Genanordnungen
Obwohl genomische Umordnungsereignisse im Laufe der Zeit zu einer Vermischung des Genomgehalts führen, lassen sich Regionen mit konservierten Ordnungen und Orientierungen in mehreren Arten identifizieren. Diese werden als Syntenieblöcke bezeichnet, in denen äquivalente Gene verschiedener Arten (Orthologe) ihre lokale genomische Nachbarschaft beibehalten haben. Genom-Entwürfe bestehen aus Genomregionen, die zu unterschiedlich langen Gerüsten zusammengesetzt sind, deren relative Reihenfolge und Ausrichtung entlang der Chromosomen jedoch in der Regel unbekannt ist. Die SIB-Forscher stellten die Hypothese auf, dass konservierte Syntenieblöcke als Grundlage für einen evolutionär orientierten Ansatz zur Ordnung und Ausrichtung von Gerüsten dienen könnten, um die derzeit fragmentierten Entwürfe zu verbessern. „Die Logik ist recht einfach“, erklärt der leitende Forscher Robert Waterhouse, SIB-Gruppenleiter am Departement für Ökologie und Evolution der Universität Lausanne: „Wenn Gene, die sich an den Enden eines Gerüsts einer Spezies befinden, Orthologe aus vielen anderen Spezies haben, die als genomische Nachbarn erhalten geblieben sind, dann legt die Evolution nahe, dass wir diese Gerüste zusammenfügen können, um diese Genpaare wieder zu vereinen.“
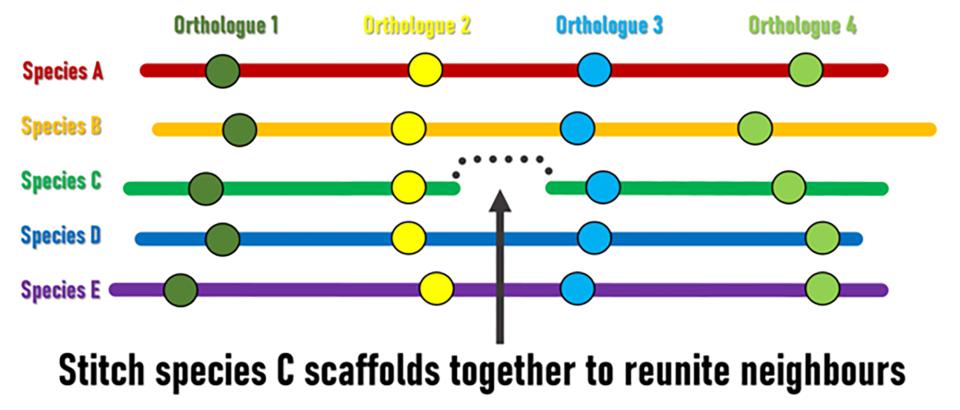
Superscaffolding und Chromosomenverankerung
Für Teilmengen der Assemblierungen integrierten die Forscher die synteniebasierten Scaffold-Adjazenzen mit zusätzlichen unterstützenden Daten aus physikalischen Kartierungsexperimenten, RNA-Sequenzierung und zusätzlichen DNA-Sequenzierungsproben. Die kombinierten Analysen ergaben 20 verbesserte Superscaffold-Assemblierungen, bei denen die Zuordnung der Scaffolds zu Chromosomen mehr als 75 % mehrerer Assemblierungen umfasste. Die Chromosomenverankerung der Scaffolds wurde für mehrere andere Assemblierungen erheblich erweitert, und für zwei Spezies wurden aktualisierte hochauflösende zytogenetische Fotokarten erstellt. Die Integration dieser verschiedenen Datensätze ermöglichte nicht nur ein verbessertes Superscaffolding, sondern diente auch als unabhängige Validierung der synteniebasierten Vorhersagen und ihrer Konsenssätze.
Die Waterhouse-Gruppe hat sich mit Forschern der George Washington University (USA) und der Simon Fraser University (Kanada) zusammengetan, um ihre drei unabhängig voneinander entwickelten Bioinformatik-Methoden, die jedoch alle auf dem gleichen Grundprinzip beruhen, zur Identifizierung solcher „reunitable neighbours” (auch als „scaffold adjacencies” bekannt) anzuwenden. Sie testeten die Leistungsfähigkeit ihrer Methoden an einem Datensatz von 21 Anopheles-Mückengenomen, darunter überwiegend fragmentierte Entwürfe von Genomsequenzierungen. Orthologe Gene, die als genomische Marker zur Definition konservierter Syntenieblöcke verwendet wurden, wurden mit Hilfe des OrthoDB -Orthologie-Abgrenzungsverfahrens, einer SwissOrthology SIB-Ressource, identifiziert .
Verbesserung der evolutionären Inferenz
Durch die Nutzung der kombinierten Erkennungsleistung der drei auf Gensyntonie basierenden Methoden konnten die Analysen Konsenssätze von Tausenden gut belegten Scaffold-Adjazenzen identifizieren, die zum Aufbau von „Superscaffolds“ (Sätzen aus miteinander verbundenen Scaffolds) verwendet wurden, was zu erheblichen Verbesserungen bei mehreren Assemblierungen führte. Zwar erfordern viele Anwendungen in der Genomforschung keine derart hochwertigen Assemblierungen, doch können Verbesserungen hinsichtlich Vollständigkeit, Kontiguität und Chromosomenverankerung oder -zuordnung die Aussagekraft und Bandbreite biologischer und evolutionärer Schlussfolgerungen aus vergleichenden Genom- oder Populationsgenetik-Analysen erheblich steigern.

Reference(s)
Waterhouse R et al. Evolutionäre Superscaffolding und Chromosomenverankerung zur Verbesserung der Genomassemblierung von Anopheles. BMC Biology 2019.






