

Le nuove tecnologie di sequenziamento del DNA e gli algoritmi di analisi delle sequenze bioinformatiche sono in grado di produrre assemblaggi di alta qualità a livello cromosomico di genomi di grandi dimensioni. Tuttavia, molte comunità di ricerca si affidano ancora ad assemblaggi provvisori "Versione 1.0" che sono frammentati, incompleti e privi di dati sulla posizione cromosomica. In uno studio pubblicato sulla rivista BMC Biology, il gruppo SIB di Robert Waterhouse (Università di Losanna) dimostra come gli approcci evolutivi basati sul confronto dei genomi possano essere utilizzati per aiutare tali assemblaggi preliminari nel loro percorso verso la realizzazione di genomi di riferimento "completi".
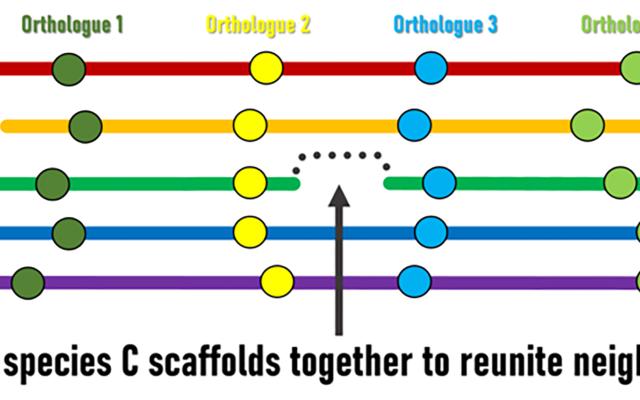
Sfruttamento delle disposizioni geniche conservate
Sebbene gli eventi di riorganizzazione genomica portino nel tempo a una rimescolanza dei contenuti del genoma, è possibile identificare regioni con ordini e orientamenti conservati in più specie. Queste sono note come blocchi di sintenia, in cui geni equivalenti in specie diverse (ortologhi) hanno mantenuto le loro vicinanze genomiche locali. I genomi provvisori sono costituiti da regioni genomiche assemblate in scaffold di diverse lunghezze, ma il loro ordine e orientamento relativi lungo i cromosomi è solitamente sconosciuto. I ricercatori del SIB hanno ipotizzato che i blocchi di sintenia conservati potessero essere utilizzati come base per un approccio guidato dall'evoluzione per ordinare e orientare gli scaffold al fine di migliorare gli assemblaggi provvisori attualmente frammentati. "La logica è piuttosto semplice", spiega il ricercatore capo Robert Waterhouse, responsabile del gruppo SIB presso il Dipartimento di Ecologia ed Evoluzione dell'Università di Losanna, "quando i geni situati alle estremità degli scaffold in una specie hanno ortologhi di molte altre specie che sono mantenuti come vicini genomici, allora l'evoluzione suggerisce che possiamo unire questi scaffold per riunire queste coppie di geni".
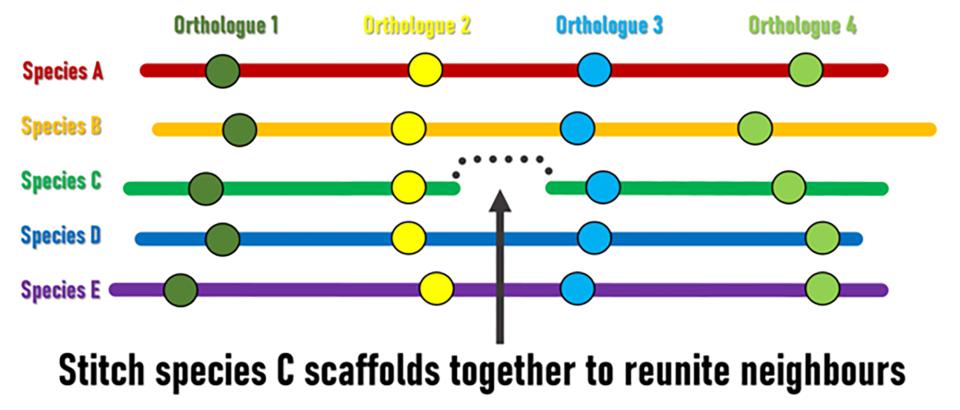
Superscaffolding e ancoraggio cromosomico
Per i sottoinsiemi degli assemblaggi, i ricercatori hanno integrato le adiacenze degli scaffold basate sulla sintenia con ulteriori dati di supporto provenienti da esperimenti di mappatura fisica, sequenziamento dell'RNA e campioni aggiuntivi di sequenziamento del DNA. Le analisi combinate hanno prodotto 20 assemblaggi superscaffolded migliorati, in cui l'assegnazione degli scaffold ai cromosomi copriva oltre il 75% di diversi assemblaggi. L'ancoraggio cromosomico degli scaffold è stato notevolmente esteso per diversi altri assemblaggi e sono state prodotte fotomappe citogenetiche ad alta risoluzione aggiornate per due specie. L'integrazione di questi diversi set di dati non solo ha consentito un superscaffolding migliorato, ma è servita anche come validazione indipendente delle previsioni basate sulla sintenia e dei loro insiemi di consenso.
Il Waterhouse Group ha collaborato con ricercatori della George Washington University (USA) e della Simon Fraser University (Canada) per applicare i loro tre metodi bioinformatici sviluppati in modo indipendente, che tuttavia utilizzano tutti lo stesso principio di base, per identificare tali "vicini riunibili" (noti anche come adiacenze di scaffold). Hanno testato le prestazioni dei loro metodi su un set di dati di 21 genomi di zanzare Anopheles, comprendente principalmente assemblaggi frammentati. I geni ortologici, utilizzati come marcatori genomici per definire i blocchi di sintenia conservati, sono stati identificati utilizzando la procedura di delineazione ortologica OrthoDB, una risorsaSwissOrthology al SIB.
Potenziare l'inferenza evolutiva
Sfruttando la potenza di rilevamento combinata dei tre metodi basati sulla sintenia genica, le analisi hanno identificato insiemi concordanti di migliaia di adiacenze di scaffold ben supportate che sono state utilizzate per costruire "superscaffold" (insiemi di scaffold cuciti insieme), con conseguenti miglioramenti sostanziali per diversi assemblaggi. Sebbene molte applicazioni nella ricerca genomica non richiedano necessariamente assemblaggi di così alta qualità, i miglioramenti in termini di completezza, contiguità e ancoraggio o assegnazione dei cromosomi possono aumentare notevolmente la potenza e l'ampiezza delle inferenze biologiche ed evolutive derivanti dalle analisi comparative del genoma o della genetica delle popolazioni.

Reference(s)
Waterhouse R et al. Superscaffolding evolutivo e ancoraggio cromosomico per migliorare gli assemblaggi del genoma dell'Anopheles. BMC Biology 2019.






