

Les nouvelles technologies de séquençage de l'ADN et les algorithmes d'analyse des séquences bioinformatiques permettent de produire des assemblages de grande qualité au niveau chromosomique de génomes volumineux. Cependant, de nombreuses communautés de recherche s'appuient encore sur des assemblages provisoires « version 1.0 » fragmentés, incomplets et dépourvus de données sur la localisation chromosomique. Dans une étude publiée dans la revue BMC Biology, le groupe SIB de Robert Waterhouse (Université de Lausanne) démontre comment les approches évolutives basées sur la comparaison de génomes peuvent être utilisées pour aider ces assemblages préliminaires à devenir des génomes de référence « finis ».
Exploitation des arrangements génétiques conservés
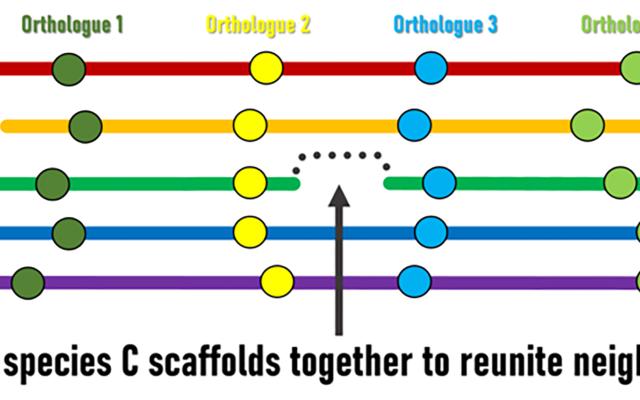
Bien que les réarrangements génomiques entraînent au fil du temps une réorganisation du contenu du génome, il est possible d'identifier des régions dont l'ordre et l'orientation sont conservés chez plusieurs espèces. Ces régions sont appelées « blocs de synténie », où des gènes équivalents chez différentes espèces (orthologues) ont conservé leur voisinage génomique local. Les génomes préliminaires sont constitués de régions génomiques assemblées en échafaudages de différentes longueurs, mais leur ordre et leur orientation relatifs le long des chromosomes sont généralement inconnus. Les chercheurs du SIB ont émis l'hypothèse que les blocs de synténie conservés pourraient servir de base à une approche guidée par l'évolution pour ordonner et orienter les échafaudages afin d'améliorer les assemblages préliminaires actuellement fragmentés. “La logique est assez simple”, explique Robert Waterhouse, chercheur principal et chef du groupe SIB au Département d’écologie et d’évolution de l’Université de Lausanne. “Lorsque des gènes situés aux extrémités des échafaudages d’une espèce ont des orthologues provenant de nombreuses autres espèces qui sont maintenus comme voisins génomiques, l’évolution suggère que nous pouvons assembler ces échafaudages pour réunir ces paires de gènes”.
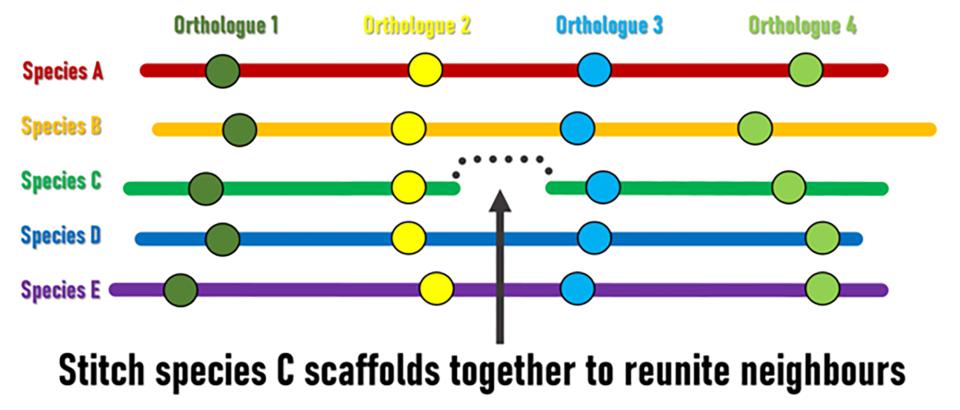
Superscaffolding et ancrage chromosomique
Pour des sous-ensembles des assemblages, les chercheurs ont intégré les adjacences des échafaudages basées sur la syntonique avec des données supplémentaires issues d'expériences de cartographie physique, du séquençage d'ARN et d'échantillons supplémentaires de séquençage d'ADN. Les analyses combinées ont permis d'obtenir 20 assemblages superscaffoldés améliorés, dans lesquels l'attribution des échafaudages aux chromosomes couvrait plus de 75 % de plusieurs assemblages. L'ancrage chromosomique des échafaudages a été considérablement étendu pour plusieurs autres assemblages, et des photomaps cytogénétiques haute résolution ont été mises à jour pour deux espèces. L'intégration de ces différents ensembles de données a non seulement permis d'améliorer le superscaffolding, mais a également servi de validation indépendante des prédictions basées sur la syntonique et de leurs ensembles consensuels.
Le groupe Waterhouse s'est associé à des chercheurs de l'université George Washington (États-Unis) et de l'université Simon Fraser (Canada) afin d'appliquer leurs trois méthodes bioinformatiques développées indépendamment, mais reposant toutes sur le même principe de base, pour identifier ces « voisins réunissables » (également appelés « adjacences de échafaudages »). Ils ont testé les performances de leurs méthodes sur un ensemble de données comprenant 21 génomes de moustiques Anopheles, dont la plupart étaient des assemblages fragmentés. Les gènes orthologues, utilisés comme marqueurs génomiques pour définir les blocs de syntonies conservées, ont été identifiés à l'aide de la procédure de délimitation orthologique OrthoDB, une ressourceSwissOrthology du SIB.
Renforcer l'inférence évolutive
En tirant parti de la puissance de détection combinée des trois méthodes basées sur la syntonéité génétique, les analyses ont permis d'identifier des ensembles consensuels de milliers d'adjacences de échafaudages bien étayées qui ont été utilisées pour construire des « superscaffolds » (ensembles d'échafaudages assemblés), ce qui a permis d'améliorer considérablement plusieurs assemblages. Bien que de nombreuses applications dans la recherche génomique ne nécessitent pas strictement des assemblages d'une telle qualité, les améliorations apportées à l'exhaustivité, à la contiguïté et à l'ancrage ou à l'attribution des chromosomes peuvent considérablement renforcer la puissance et l'étendue des inférences biologiques et évolutives issues de la génomique comparative ou des analyses de génétique des populations.

Reference(s)
Waterhouse R et al. Super-échafaudage évolutif et ancrage chromosomique pour améliorer l'assemblage du génome d'Anopheles. BMC Biology 2019.






