


SIB is a leading organization in biological and biomedical data science. It maximizes the impact of, and investments in scientific projects by making data FAIR (Findable, Accessible, Interoperable and Reusable), provides long-term access to leading Swiss biodata resources, and is a high-quality partner enabling innovation in academia, hospitals and industry. It does this through its complementary pillars on coordination, open databases and software tools, and centre of excellence. Discover below who we are, our vision, mission and what is bioinformatics. Also find out about our history and impact over the years as data scientists for life, and sign up for our activity report.
In a nutshell
SIB is an internationally recognized non-profit organization, dedicated to biological and biomedical data science.
Our data scientists are passionate about creating knowledge and solving complex questions in many fields, from biodiversity and evolution to medicine. They provide essential databases and software platforms as well as bioinformatics expertise and services to academic, clinical, and industry groups.
SIB federates the Swiss bioinformatics community of some 900 scientists, encouraging collaboration and knowledge sharing.
The institute contributes to keeping Switzerland at the forefront of innovation by fostering progress in biological research and enhancing health.
Our vision
At SIB, we know that expertise in life science data is key to solving many of the world’s most pressing challenges. By unlocking the potential of biological and biomedical data, we aim to generate knowledge and innovate for a better future.
Our mission
Our mission is to push the boundaries of data science through in-depth knowledge of biological data, cutting-edge technologies and interdisciplinary collaborations.
- We provide researchers and clinicians with outstanding bioinformatics resources, services and training to accelerate innovation in many fields, from medicine, health and agriculture to the preservation of biodiversity and the environment.
- We represent and federate Swiss bioinformatics. By fostering a culture of scientific excellence and collaboration, we contribute to ensuring Switzerland remains one of the most innovative countries in the world.
Our activity report: the SIB Profile
Our activity report is published every year. In the SIB Profile, you will discover our institution and its functioning, yearly highlights, and focus chapters to dive into selected hot topics, from machine learning to biodiversity.
SIB Profile 2025 / Activity Report 2024
Focus topics: Preparing for the next epidemic / The AI revolution continues
Tätigkeitsbericht 2025 – Auszug
Biologische Datenwissenschaft treibt Schweizer Innovation.
Rapport d’activité 2025 – extrait
La science des données biologiques, moteur de l’innovation suisse.

SIB Profile 2024 / Activity Report 2023
Focus topics: A central role for Switzerland’s standing in life science / Generative AI / One Health

SIB Profile 2023 / Activity Report 2022
Focus topics: Biodiversity / Open research data

SIB Profile 2022 / Activity Report 2021
Focus topics: Infrastructure solutions / International

SIB Profile 2021 / Activity Report 2020
Focus topics: COVID-19 / Machine learning

SIB Profile 2020 / Activity Report 2019
Focus topics: Health data / Open knowledge
Bioinformatics: a definition
Life scientists and clinicians have long tried to assemble data and evidence to find the right answers to fundamental questions. Today, however, there is no shortage of data, and we find ourselves with a different issue. New technologies are producing data at an unprecedented speed, and in such quantity and variety that they can no longer be interpreted by the human mind alone.
Enter bioinformatics.
Bioinformatics is the application of computer technology to better understand and effectively use biological and biomedical data. It is the discipline that stores, analyses and interprets the big data generated by life-science experiments, or collected in a clinical context. This multidisciplinary field is driven by experts from a variety of backgrounds: biologists, computer scientists, mathematicians, statisticians and physicists.

- Databases for storing, retrieving and organizing curated information to maximise the value of biological data
- Software tools for modelling, visualizing, interpreting and comparing biological data
- Analysis of complex datasets using novel statistical approaches or machine learning techniques
- Research harnessing computational methods in a wide variety of biological fields to develop solutions in diverse areas, from agriculture to precision medicine
- Computing and storage infrastructure to process and safeguard large amounts of biological data
Building on long-term expertise to fight pandemics
Much of the infrastructure and expertise developed over many years at SIB came into the spotlight with the recent pandemic of SARS-CoV-2. Among the SIB Resources, V-pipe is used, for instance, by international researchers to detect novel variants in wastewater, while Nextstrain enables real-time tracking of virus evolution, whether SARS-CoV-2, Ebola or Zika. Several SIB Group Leaders are also active members of the Scientific Advisory Panel to advise the government on mitigating COVID-19. Finally, the Swiss Pathogen Surveillance Platform (SPSP) was repurposed to act as the Swiss SARS-COV-2 Data hub.
Our history: leading Swiss bioinformatics for over 25 years
It all began in 1998, with an association of like-minded scientists with a common passion: to ensure the progress of life science research in Switzerland.
SIB has now grown into a major organization with far-reaching impact that can be relied on for professional services. As well as equipping researchers and policymakers to fight pandemics, supporting the development of personalized health and working towards a better environment for all, SIB also made substantial contributions to fostering research data quality, integrity and reproducibility. This is done for instance through fostering open research data, providing essential infrastructure to support advances in the life sciences, offering bioinformatics training to promote good research practices, and recognizing and encouraging excellence through its recognition initiatives.
Our evolution in a few figures
SIB has seen extraordinary growth over its 25 years of existence, with today 88 groups and 900 members spanning Switzerland who have authored over 4,000 publications.
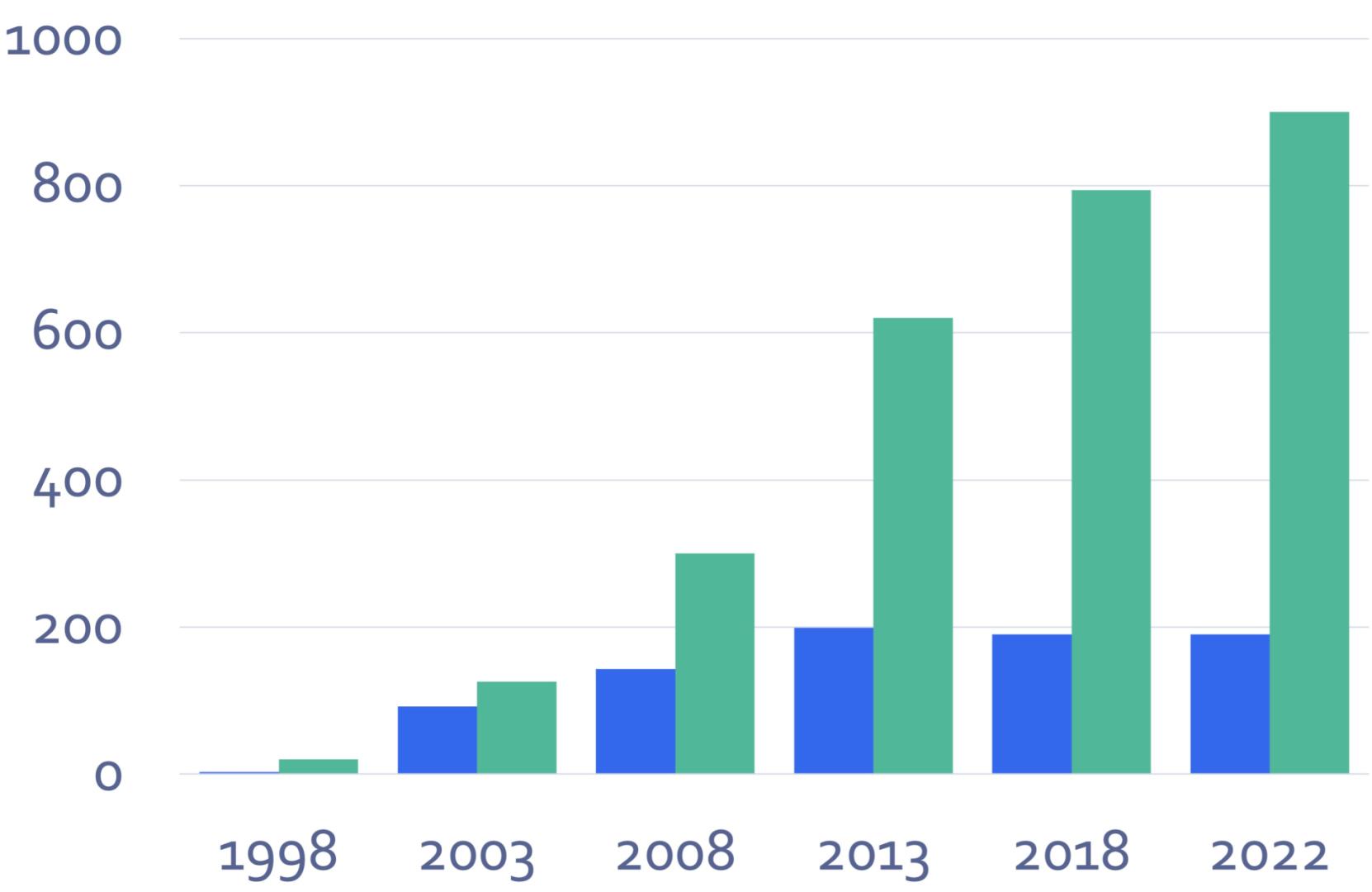
The growth in members (green) and employees (blue)
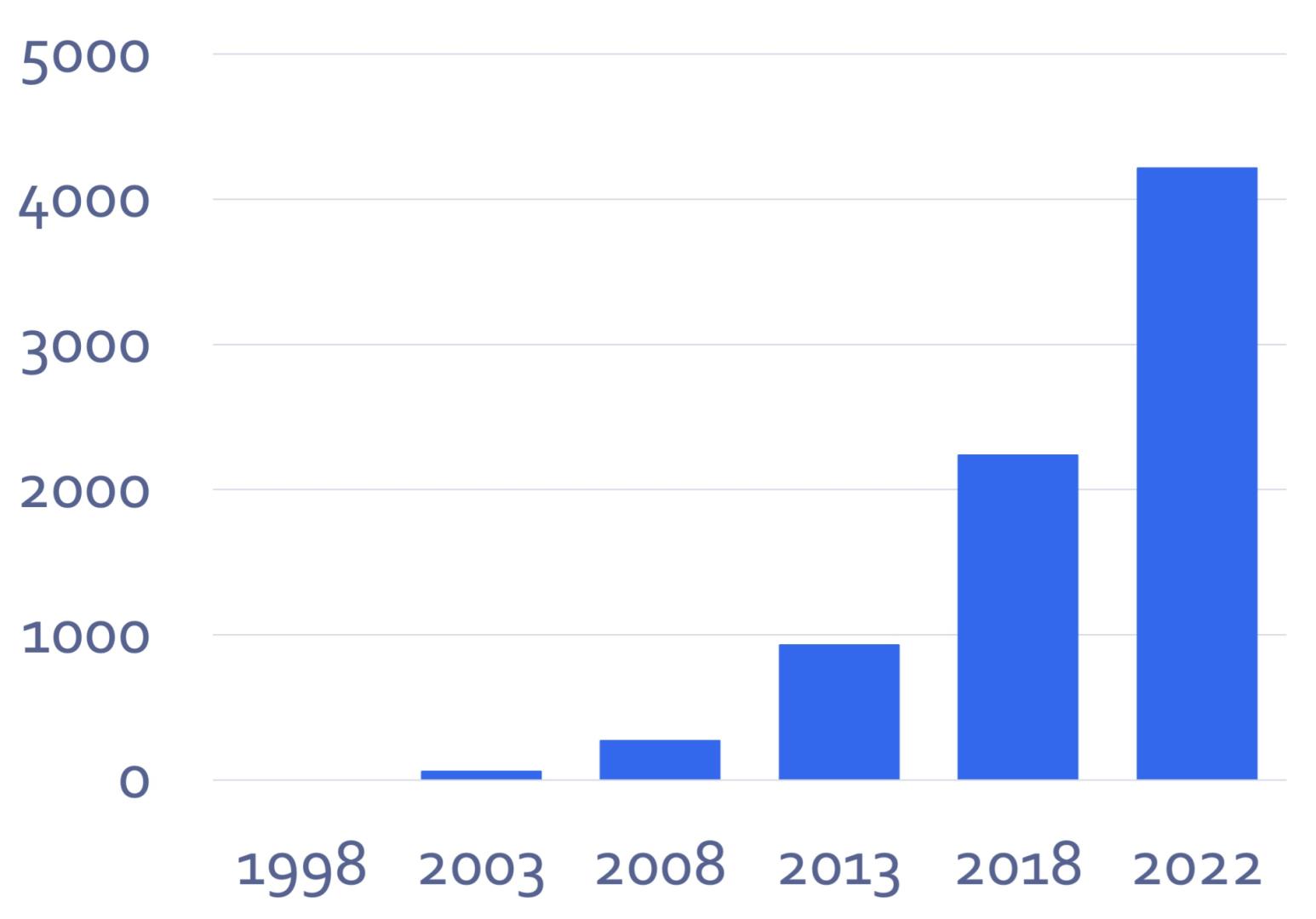
The cumulative number of publications with an author from SIB
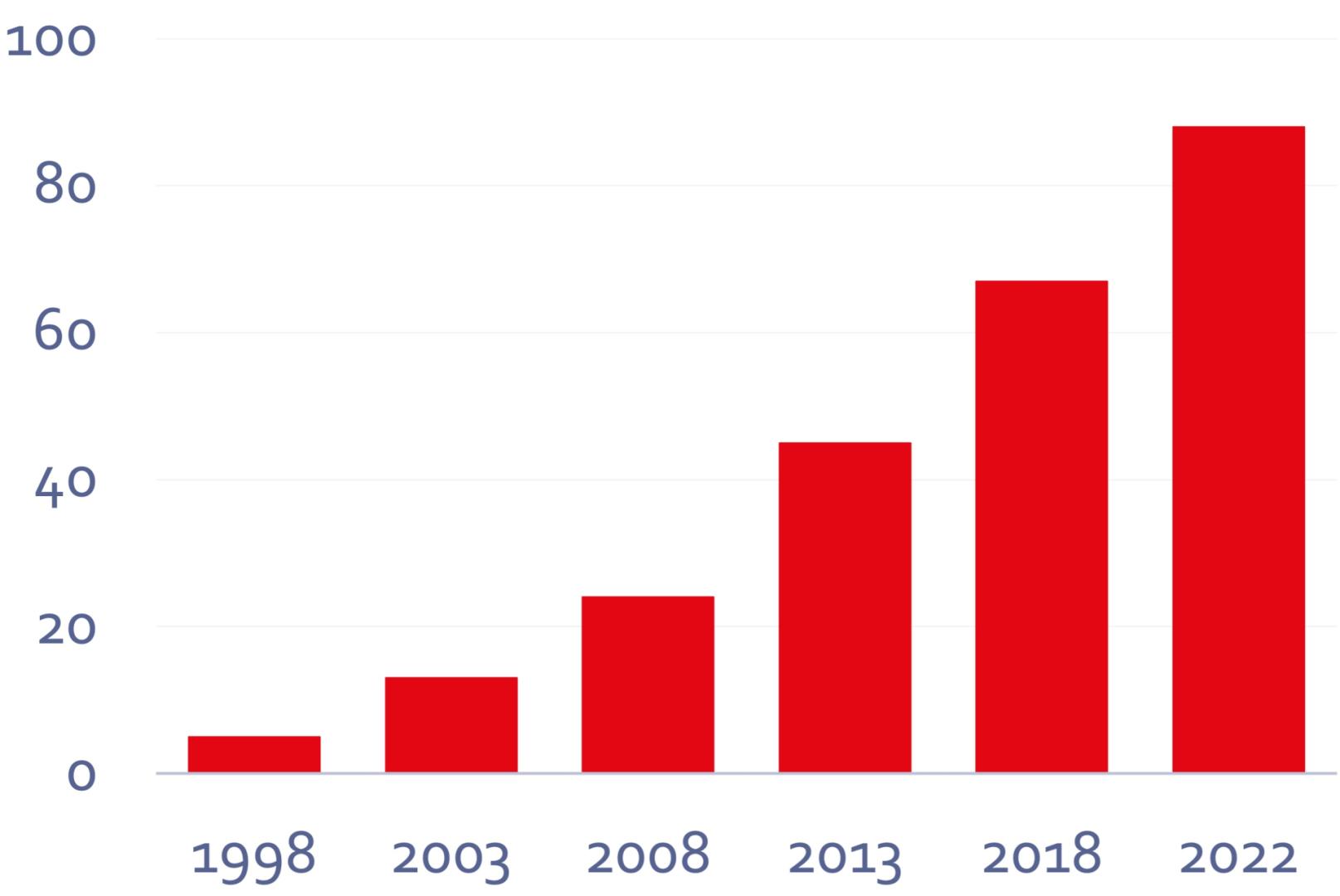
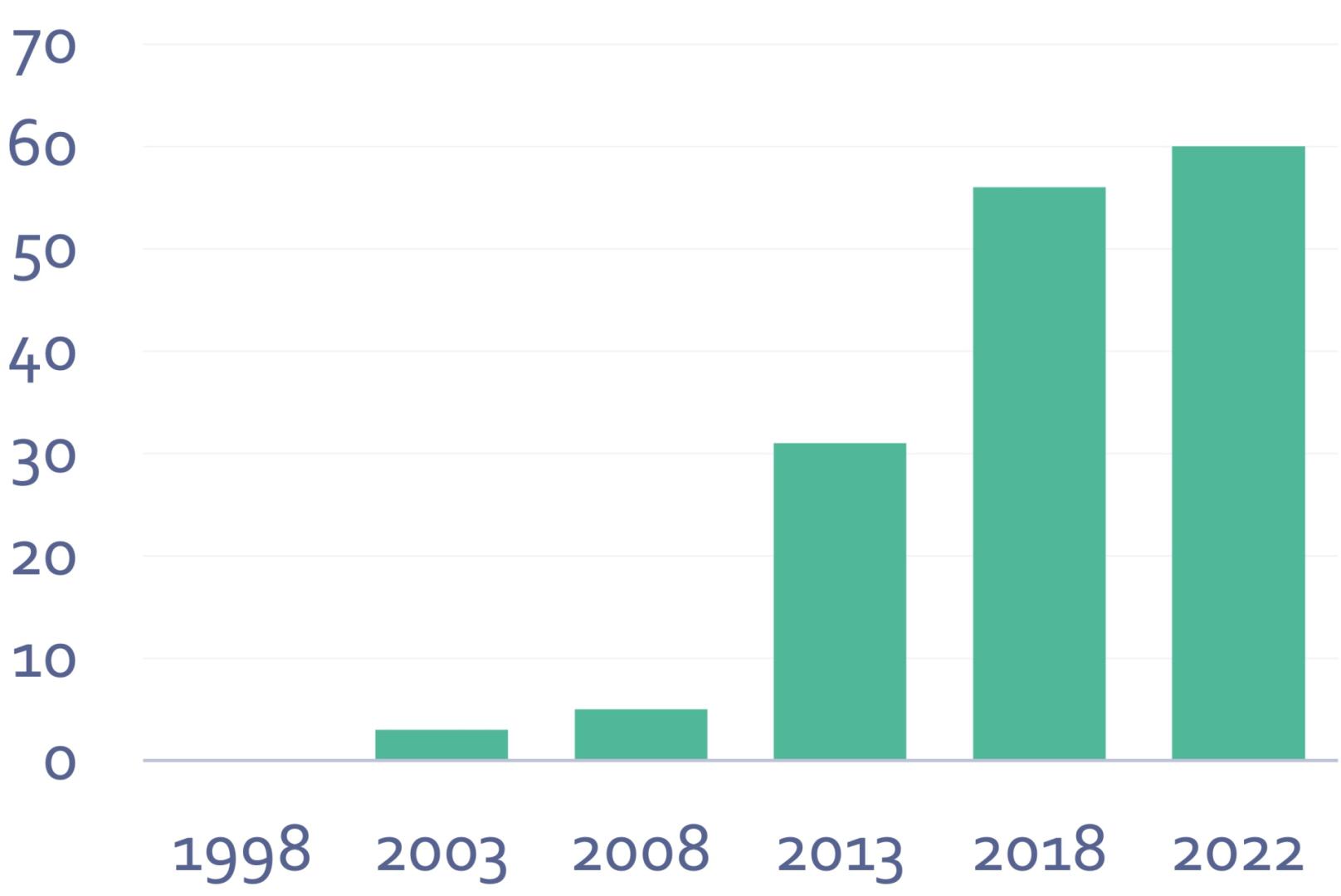
The number of courses offered by our Training group
Our brand identity
A distinct, engaging and meaningful brand identity is a key asset for an organization’s positioning and strategy.
Its distinct elements thus aim to:
- reinforce our position as a leading organization for biological and biomedical data science and innovation, and as a professional and reliable partner
- to reflect the diversity of domains and competences present at SIB
- to provide a unified and distinctive banner for Swiss bioinformatics on the national and international levels.
More about our new website and rebranding
Our name
When introducing the name of the institute for the first time in a text, it should be written in full as shown below:
- SIB Swiss Institute of Bioinformatics
- SIB Institut Suisse de Bioinformatique
- SIB Istituto Svizzero di Bioinformatica
- SIB Schweizerisches Institut für Bioinformatik
The abbreviation SIB may be used after this first occurrence.
Our logo
The Swiss flag and colours show the Swiss origin and the related excellence and reliability, the history of the institute, its federal organization. Bioinformatics, data and computerized science are illustrated by the computer screen shape of the logo. The molecule represents life sciences and health, uniting forces, connectivity and networking.
Our logo is under the creative commons license CC BY-NC-ND 4.0
Download:
Full logo, transparent background (.png)
Full logo, white background (.jpg)
Emblem, .png
Emblem, .jpg
{kind=link}
{kind=link}
{kind=link}
{kind=link}
For any use of our logo by people or institutions outside SIB’s community, and for any questions regarding its use and other branding aspects (templates, guidelines), please contact SIB Communication and Scientific events.
Graphical environment
Our activities are at the cross-road between cutting-edge technologies and human expertise. They also impact life and society across a range of domains, such as personalized medicine or biodiversity. Our brand identity reflects these different facets:
- modern and impactful colours ranging from warm pink to electric blue;
- graphic elements that play around the themes of digitalization, data integration, biology, big data.

Our typefaces
The Cabinet Grotesk typeface used for our headlines and highlights has a contemporary design and conveys an idea of precision and analysis.
The Inter UI typeface is used for our body texts. It was selected for its readability and modern, innovative look.